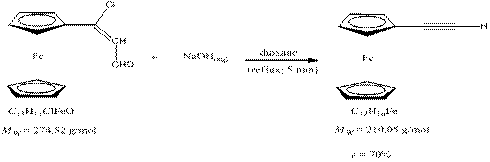Română English
Bilateral ProjectRomania-Ungaria
|
||||||||||||
|
General method of synthesis some pyrazoles The compound 10-methyl-3-[1-methyil-3-(phenil)-1H-pyrazole-5-yl]-10H-phenothiazine 2a is a yellow powder. Because, behind the reaction were resulted more isomers, the purification of this compound is still in progress . The compound 10-(n-buthyl)-3-[1-methyl-3-(ferrocenyl)-1H-pyrazole-5-yl]-10H-phenothiazine 2b appear as red cristals which have m.t.=77-790C. Elemental analysis for C30H29FeN3S (519), calculated: C 69,4; H 5,6; N 8,1; S 6,2% 1H-NMR, CDCl3, 500MHz: The compound 10-ethyl-3,7-bis-(1-methyl-5-phenil-1H-pyrazole-3-yl)-10H-phenothiazine 4a was obtained in this way: to 0,0015 mol (0,73 g) bis-chalcone 3a which was suspended in 50 ml acetic acid was added 0,006 mol (0,272 g; d= 0,88 g/cm3) methylhidrazine. The reaction mixture was kept to reflux 8h. After that it was concentrated to 1/5 from initial volume, by vacuum distilation and than was added 10 ml distilled water. The obtained precipitate was filtrated, was washed with 2x10cm3 water, than it was suspended in 20 ml methanol in which was introduced air time of 1-1,5h. After removing the solvent, the isolation of the compound 4a. was realized by column chromathography using as eluent toluen and than recristalisation from methanol-n-hexane (2:1) when was obtained white-yellow cristals. Elemental analysis for C33H27N5S (525), calculated: C 75,4; H 5,2; N 13,3; S 6,1% The compound 8 appears as yellow cristals. Yield η =35-40%.
Bibliography:
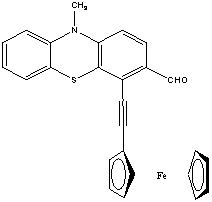
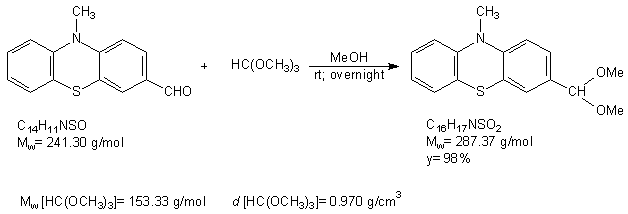
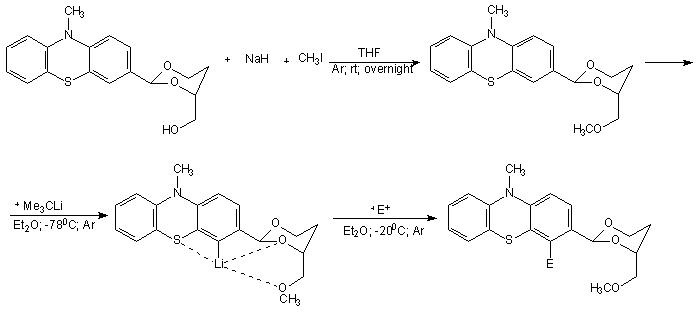
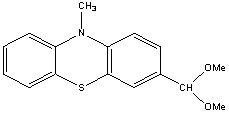
Research report of the Hungarian researchers Kiss Kolos and Gyomore Adam They were proposed two research topics. A. Gyomore has to protect the formyl group of the 10-methyl-10H-phenothiazine-3-carbaldehyde with (±) 1,2,4 –butanetriol for lithiate this substance in the position number four. K. Kiss has to prepare ethnylferrocene via acethylferrocene and 3-chloro-3-ferrocenylacrylaldehyde. Ethynilferrocene can links with for example 4-iodo-3-(4-(methoxymethyl)-1,3-dioxan-2-yl)-10-methyl-10H-phenothiazine, to form the following compound after the acidic hydrolisis: Procedure The formylating agent was prepared from. 5 cm3 POCl3 (53.64 mmol) added to 10 cm3 (129.16 mmol) abs. DMF at 0 oC. It mixed until it cooled back 0oC. 10 g (46.90 mmol) 10-methyl-10H-phenothiazine suspended in 10 cm3 abs. DMF and the suspension added to the solution. The remained substance was washed into the solution with 10+5+5 cm3 abs. DMF. This solution was heated to 104 oC and mixed for 4 hours. After the heating the mixture quenched with icy water. Yellowish precipitate was observed. Than solid NaHCO3 added to this mixture until the pH was 7.5. Next day the solution extracted with the following amounts of toluene: 250-150-3 x 50 cm3. The extract was dried on anhydrous Na2SO4, was filtered and the solvent was removed. The material crystallized with small amount of ethanol and water. The crude product was a yellowish powder like solid. Next day it was chromatographed on silicagel used toluene as eluent. The first fraction was 10-methyl-10H-phenothiazine and the fourth one was 10-methyl-10H-phenothiazine-3-carbaldehyde. The product is yellow crystalline solid. m(product) = 3.22 g (13.34 mmol) The next step was the protecting of formyl group. The protecting group chosen was the 4-(methoxymethyl)-1,3-dioxane group In the second step A. Gyomore converted the formyl group of the 3-formyl-10-methyl-10H-phenothiazine to dimethoximethyl group. Methanol was dried with sodium and distillated. 1.13 g (4.683 mmol) 10-methyl-10H-phenothiazine-3-carbaldehyde was added to a 50 ml flask. The compound was dissolved in 20 ml methanol. Yellowish suspension was observed. 20 ml trimethoxymethane was added to the suspension. One drop concentrated (96%) sulphuric acid was added to the suspension. The suspension immediately became a solution. The solution was stirred overnight at room temperature. Next day it was added to 200 ml saturated NaHCO3 solution and extracted with dichloromethane until the dichloromethane was colorless. The yellowish extract was dried on CaCl2 and the solvent was removed with vacuum evaporation. The material left crystallize overnight resulting a yellowish brown solid compound. m(product) = 1.32 g (4.593 mmol) In the third step the dimethoximethyl group was substituted by a 4-(hydroximethyl)-1,3-dioxane group.
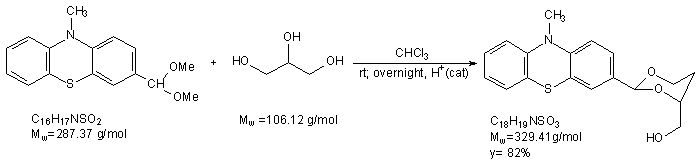
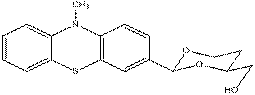
A toluenic suspension of 0.54 g (±) 1,2,4-butanetriol was dried by azeotrop distillation (a suspension was made in toluene, than toluene evaporated). This procedure was repeated two more times. The triol was put to an exicator and left it overnight over CaCl2. 4.42 g 3Å molecular sieves pellets werw activated in a drying tube at 143 oC for 10 h. A suspension of the triol with 6 ml chloroform was added the activated molecular sieves, than 0.05 g (S) – camphorsulfonic acid was added to the suspension. 1.32 g (4.593 mmol) acetal was washed with 10 ml chloroform to the mixture. The mixture left stirring overnight with CaCl2 charged tube. Next day the mixture was filtered and the molecular sieves pellets was washed with dichloromethane. The solution was filtered on cellite, dried over K2CO3 and m(product) = 1,24 g (3.764 mmol) The following steps will be performed at ELTE University. Project from the synthesized (2-(10-methyl-10H-phenothiazin-7-yl)-1,3-dioxan-4-yl)methanol In the next step A. Gyomore has to methylate the OH – group. After methylation the compound is lithiable in the fourth position with Me3CLi in dry diethylether. The lithium salt can be quenched with some electrophiles.
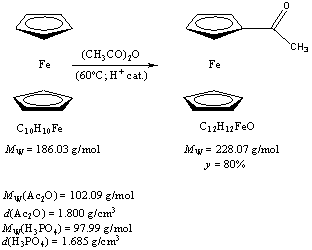
K. Kiss work The research topic proposed was the synthesis of ethyinilferrocene from ferrocene and ethynilferrocene linkable iodoaryl and bromoaryl compounds with palladium catalyst, like 4-iodo-3-(4-(methoxymethyl)-1,3-dioxan-2-yl)-10-methyl-10H-phenothiazine. Firstly K. Kolos prepared acetylferrocene from ferrocene according to a literature procedure5,6.
Procedure K. Kolos added 31 ml Ac2O and 6.50 g (34.94 mmol) to a 100 ml flask. K. Kolos got a suspension. After K. Kolos suspended the ferrocene in aceticacidanhydride, the suspension was heated to 60oC. Concentrated phosphoric acid was added to this mixture and in the case of addition the color of suspension was changed brownish. After 30 minutes stirring, the reaction mixture was quenched to ice. Orange color precipitate was eliminated from the solution. After the ice melted, Na2CO3 was added to the mixture until the pH is 5. Than saturated Na2CO3 solution was added to the same mixture until the pH ~ 6-7. The precipitate filtered with Büchner – funnel and washed with cold water. After the drying in exicator he got 6.40 g (28.06 mmol) product.
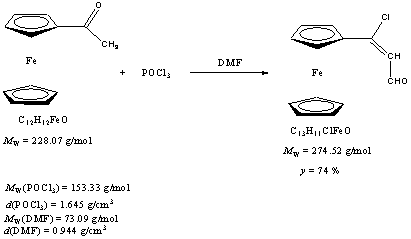
Procedure 9 ml abs. DMF and 2.5 ml POCl3 was mixed in a two necked 250 ml flask at 0oC. At this temperature the DMF solution of acetylferrocene was added to the solution via dropwise for 20 minutes. After that the solution was stirred more 15 minutes at 0oC. After the quarter hour ticked away the solution was allowed to warm room temperature and stirred for 4 hours. The color of the solution became indigo blue. 120 ml 20 w% 0oC sodiumacetate solution was added and it was stirred overnight. Next day the mixture was extracted with chloroform and the organic layer washed with water three times. The solution dried over CaCl2. After drying the solution filtered and the solvent evaporated. The dark red oil was crystallized with ciklohexane. Finally he got dark red crystalline solid. m(product) = 1.78 g (6.338 mmol).
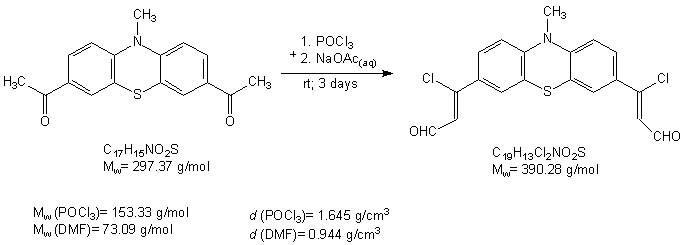
Procedure 0.48 g (1.734 mmol) of 3-chloro-3-ferrocenylacrylaldehydewas added to 15 ml abs. 1,4 –dioxane in a 50 ml three necked flask under argon atmosphere. After that the mixture was heated to reflux, 10 ml 0.5 M NaOH solution was suddenly added to the mixture. After 5 min reflux, the mixture was added to 50 ml cold water. The crude product formed as yellowish precipitate from water. 0.1 M HCl solution was added to this system until pH=5. The precipitate was filtered on and washed with water. The crude pruduct was dried with vacuum pump for 2 hours and cromatographed on neutral aluminiumoxide with petrolether 40/60 eluent. The product is a bright yellow powder like material. m(product) = 0.26 g (1.238 mmol) K. Kolos and A. Gyomore have tried to prepare 3,3'-(10-methyl-10H-phenothiazine-3,7-diyl)bis(3-chloroacrylaldehyde) with Vielsmayer formylation from 1,1'-(10-methyl-10H-phenothiazine-3,7-diyl)diethanone.
Procedure Kolos and Adam mixed 1.9 cm3 (0.0201 mmol) POCl3 and 5.8 cm3 abs. DMF in a 250 ml two necked flask at 0oC. 1 g (2.562 mmol) 1,1'-(10-methyl-10H-phenothiazine-3,7-diyl)diethanone suspended in 30 ml DMF. The formylating agent added to the suspension via dropwise. The mixture stirred more 15 min at 0oC. Than the mixture allowed to warm room temperature and stirred more 20 hours. Next day the it quenched with100 cm3 20 w% sodiumacetate solution. The mixture stirred for 72 hours. After it the mixture extracted with chloroform, the organic layer washed with water three times and dried over anhydrous calciumchloride. The solution filtered and concentrated by rotary vacuumevaporator. The crude product crystallized with normal heptane and filtered with Hirsch funnel. Adam and Kolos haven`t found an eluent yet which can separate the compounds. m(crude product) = 1.14 g
|