Introduction
Theoretical calculations at the HF/6-31G(d,p) level of theory were conducted on a large series of calix[n]arenes and calix[n]arenes (n = 4, 5, 6, 8), some of which were employed as guest molecules in subsequent calculations. Different substituent groups were employed on both the upper and lower rims to analyze their influence on its binding properties. A conformational study was performed on various complexes of calix[4]arene tetrasulfonate with alkaline cations and one neutral molecule with biological activity, hereafter referred to as GTP.
The theoretical study conducted included geometry optimizations, vibrational frequencies analysis and Basis Set Superposition Errors in order to assess the strength of the host-guest interactions. In selected cases the NBO methodology was used to assess the bonding properties of our systems, also the NBO deletion methodology was employed to investigate the bonding strength between hosts and guests.
Results
Geometry optimizations were carried out for all compounds, leading to minima on the PES which was confirmed by the absence of imaginary frequencies after the vibrational frequency analysis. The aforementioned optimizations were carried out by increasing the level of theory in the following order:
MM -> PM3 -> HF/3-21G** -> HF/6-31G**
No DFT methods were selected so far since it is known that the π-π interactions play an important role in the stability of complexes with other aromatic moieties and such methods lack the dispersion terms needed for their proper description.
Circular H-bonding strength on calix[4]arene tetrasulfonate was evaluated through the calculation of the exchange barrier by means of the concerted reorientation of the OH groups. Three different approaches were employed: {1} Rigid potential energy surface scan, only the values pertinent dihedral angles were changed while all other variables were kept intact; {2} Fully relaxed scan, all geometry variables were optimized at every step; {3} Partially relaxed scan, scanned dihedral angles were kept frozen at each given value of the scan. The values for the energies of the scans are given in table 1, the one labeled as partially relaxed is the more realistic in terms of the dynamical physical process.
Table 1 Energy barriers calculated at the HF/6-31G(d,p) level of theory, for the concerted OH bond reorientation in the lower rim of calix[4]arene tetrasulfonat
|
"rigid scan" |
"Fully Relaxed" |
"Partially Relaxed" |
Bariera energetica |
429.3578 |
70.7834 |
180.9034 |
Table 2 summarizes the results of our NBO deletion analysis for the circular H-bonding system in the alkaline complexes of calix[4]arene tetrasulfonate. These values exhibit a relation between the location of the guest ion in the calixarene and the disruption of the circular H-bond on the lower rim. The ions locations are as follow: Li+ lays on the plane of the lower rim; Na+ stays in the middle of the cavity while Cs+ is located above the upper rim coordinated to the sulfonato groups.
The orientation of the O-H bonds in the Li+ complex is changed from the free host in such a way that the circular H-bond is totally disrupted; suggesting thus that the lone pairs on O atoms are now coordinating towards the ionic center. Therefore, elimination of these aforementioned elements on the Fock matrix will yield little difference in energy compared to the one obtained on the free host. As for the Cs+ complex, its influence on the geometry of the lower rim’s H-bond is very low, hence causing a minor disruption on its contribution to the overall energy. The case of the Na+ complex is interesting since after the geometry optimization two sulfonato groups form a Hydrogen bond closing the upper rim of the cavity and thus enclosing the ion inside. This trapping effect is advantageous as to making a sensitive agent. In order to further eliminate the influence of the ions on the interactions of the lower rim the same calculations were performed in the absence of the corresponding ion while keeping the previously obtained equilibrium geometry for the calixarene.
Table 2 NBO Deletion analysis at the HF/6-31G(d,p) level of theory from the circular H- bonding on the lower rim of calix[4]arene sulfonate
|
fara ion |
Li+ |
Na+ |
Cs+ |
schimbarea energiei in prezenta ionului |
127.9878 |
11.6835 |
16.8846 |
45.3660 |
schimbarea in energie in absenta ionului [kJ/mol] |
127.9878 |
15.4458 |
20.7887 |
47.3903 |
influenta ionica in procente [%] |
-- |
32.20 |
23.12 |
4.46 |
It can be observed that elimination of the Li+ ion increases the change in energy since the lone pairs of the Oxygen atoms in the lower rim are involved in the coordination of the ion and not in H-bonding. There is only a slight change for the Cs+ complex.
BSSE energies for the same complexes at their equilibrium conformations were also computed as a way to assess the strength of the interactions between host and guest. The obtained values are listed in table 3.
Table 3 BSSE calculations using the counterpoise method are reported in the following table.
Ion |
Li+ |
Na+ |
Cs+ |
energia BSSE |
18.5198 |
19.5790 |
9.6549 |
The molecule 3-phenyl-3a,8b-dihydro-1H-[1]benzofuro[3,2-c]pyrazole (figure 1) has proven to be a useful biological agent as a tyrosine kinase inhibitor in leukemia cells. We selected this molecule as a guest since it has a low dipole moment and hence a low water solubility, making it suitable to be delivered within a more soluble carrier.
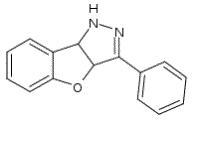
Figura 1. Structura chimica a GTP
Insertion of GTP in different calixarenes was done in two ways: (a) according to the coupling of dipole moments and (b) in terms of shape fit; in this latter case two other different fittings were possible: (b1) with the phenyl group pointing towards the lower cavity along the main axis of the calixarene and (b2) with the benzofurane ring pointing in the aforementioned direction.
Table 4 BSSE calculations on GTP complexes with different kinds of calix[n]arenes in different orientations
Compus |
n = |
Substituentii din upper rim |
Substituentii din lower rim |
Orientarea |
Energia relativa |
energia BSSE [kJ/mol] |
|
1 |
4 |
EtO- |
OH |
(b1) |
- |
8.9442 |
|
(b2) |
-3.2200 |
11.1227 |
|||||
2 |
5 |
EtO- |
Me |
(b1) |
-383.3481 |
50.0533 |
|
(b2) |
- |
12.0393 |
|||||
3 |
5 |
EtO- |
OH |
(b1) |
- |
10.8957 |
|
(b2) |
-22.7924 |
14.5647 |
|||||
4 |
6 |
SO3H |
OH |
(b1) |
- |
32.8376 |
|
(b2) |
-18.0235 |
9.8784 |
|||||
In general the BSSE is larger for (b2) orientation since there is a larger overlap between the three fused rings and the aromatic rings that constitute the calixarene skeleton. In addition the O and N atoms may form H-bonds with some of the functional groups in the upper rim.
The case of compound 4 is singular since a large 32.8 kJ/mol BSSE energy is obtained for the orientation (b1) while the orientation (b2) seems to be the most stable. Aside from this compound all others exhibit the expected trends and the (b2) orientation (benzofurane ring inside the cavity) yields the most stable conformation, except in compound 2 where a true comparison is dubious since in the (b1) fitting orientation the GTP molecule is fully encapsulated by the cavity.
Conclusions
In the case of GTP the interactions with the guest are mainly dominated by dispersion forces and p-p stacking interactions. In some instances an occasional and accidental H-bond may help keeping the guest inside the cavity whenever the substituents on the upper rim are capable of forming them.
Calix-ions interactions are mostly driven by fitting than by ionic-dipole interactions. This can be concluded through careful analysis of the BSSE results between the Li and the Na complex. The Cs complex constitutes a different class since the same basis set used for the other two was not available for Cs.
Disruption of the circular H bonding in the lower rim of calixarenes by the presence of cationic guests is caused by its location and not by its ionic potential. Hence the Cs complex has the lowest deletion change in energy since the cation is lays on the upper rim as compared to the Na complex in which the cation is completely immersed in the cavity. The Li complex completely looses the circular H bonding since the cation lays on the plane of the lower rim.
______________________ |
______________________ |
______________________ |
Dr. Joaquin Barroso-Flores |
Dr. Ioan Silaghi Dumitrescu |
Dr. Kunsagi-Mate Sandor |