Radu Silaghi-Dumitrescu
Activation of molecular oxygen and
of peroxides by hemoproteins and by non-heme iron systems
Proteins such as cytochrome P450 (P450), horseradish peroxidase
(HRP), hemoglobin and catalase are all known to bind
and/or reduce (activate) dioxygen at their heme active sites, in the ferrous form. A brief overview of
these processes is given below. While the thiolate-ligated ferrous heme of P450 binds dioxygen and
promotes O-O bond cleavage, the ferrous histidine-ligated heme
of hemoglobin only binds dioxygen in a reversible
manner. Thiolate- as well as histidine-ligated active sites are
also known to form ferric-hydroperoxo complexes. In
HRP the hydroperoxo complex decays via
proton-assisted heterolytic cleavage of the O-O bond
to yield water and a

Physiologically-relevant reactions of active
site hemes with O2 and/or H2O2. “S” denotes an organic
substrate molecule. “X” may be a protein-derived cysteinate or histidine
ligand.
Our interest has been in defining the properties and
reactivity of the transient adducts depicted in the Figure above (nature of
ferrous-dioxygen bonding, nature of ferric-peroxo interaction, electronic structure and protonation
state of ferryl, i.e. Compounds I and II). Traditional manners of cleaving the
O-O bond in metal-hydroperoxo complexes, and especially
in Fe(III)-OOH, have been either proton-unassisted, or
proton-assisted involving an Fe(III)-O-OH2 intermediate/transition
state. However, an alternative mechanism has been proposed by Li et al, based
on computations and involving an Fe(III)-O(H)-OH intermediate that undergoes
O-O bond cleavage directly, avoiding the Fe(III)-O-OH2 species. The
Figure below illustrates two examples of particularly facile O-O bond
activation in Fe(III)-OH-OH adducts computed by us.


Upper panel: potential energy surface, following O-O
bond elongation in a Fe(III)-OH-OH model of bleomycin (structure shown in inset, left lower corner).
Geometries were optimized with the HO-OH distance constrained to values
indicated in the plots, starting from the equilibrium value of 1.51 Å. Energy
differences are plotted for each model, with the energy of the equilibrium
structure (far left side of the plot) taken as reference for each model. At an
O-O distance of 2.2 Å, both protons originating from the HOOH ligand were found
on the leaving oxygen atom, yielding the structure depicted in the inset, upper
right corner. Further optimization without any geometry constraints led to a
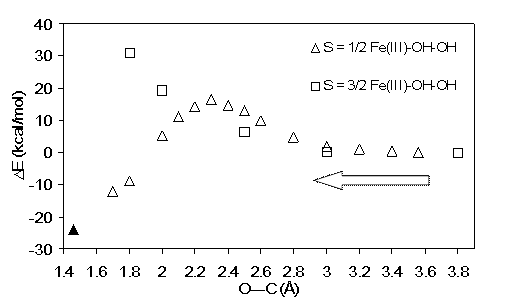
final O-O distance of ~2.7Å (rightmost data point in the plot). Lower panel: potential energy surface,
following meso-hydroxylation reactions in a Fe(III)-H2O2; X = imidazole, i.e. a
possible mechanism for the reaction performed at the active site of heme oxygenase. Geometries were
optimized with the O---C distances (shown as dotted lines in the reaction
scheme) constrained to values indicated in the plots, starting from the
equilibrium geometry with C---O ~3.8 Å, and shortening this distance by amounts
indicated in the plot. The leftmost data point (solid triangle, S=1/2 model) was
obtained by geometry optimization after lifting all geometry constraints.
Energy differences are plotted for each model, with the energy of the
equilibrium structure (far right side of the plot) taken as reference. No O-O
bond cleavage was found to occur for the equivalent S=3/2 Fe(III)-OH-OH
model under the conditions used here (instead, at the left-most point of the
plot, O---C = 1.8 Å, the Fe-O bond was irrevocably broken)