Radu Silaghi-Dumitrescu
Nitrite reduction by
nitrite reductases and related model systems, and the role of linkage isomerism
Reduction of nitrite to nitric oxide is essential in
certain living species, and has in fact recently been proposed to be an important
secondary function of hemoglobin in humans (cf
Gladwin, NATURE - CHEM BIOL 2005, 245). In vivo, nitrite reduction is
accomplished by metalloenzymes, involving direct metal-nitrite coordination.
Recently, we proposed that linkage nitro/nitrito isomerism
is an essential part of the mechanism in copper and in heme
d1-containing nitrite reductases (cf Silaghi, INORG CHEM 2004, 256). Recent experimental work
has also shown involvement of linkage isomerism in catalytic reduction of
nitrite by free hemes and related small complexes in
solution.
Under anaerobic conditions, many microorganisms can
sustain growth by using nitrate as respiratory terminal electron acceptor. Much
like dioxygen, partially reduced products of nitrate
may be toxic to living cells. While respiratory dioxygen
reduction delivers all four electrons in one single step (O2 + 4H+
+4e- -> 2H2O), respiratory nitrate reduction in
bacteria and archaea is more complex, proceeding
stepwise on more than one possible pathway. Bacteria in fact exhibit a wide
range of metabolic reactions with various oxides of nitrogen, sometimes
referred to as the bacterial nitrogen cycle, which is diagrammed in Figure 1.
[Richardson et al, Curr. Opin. Chem. Biol. 1999, 207]

Figure 1.
The bacterial nitrogen cycle. The oxidation states of
nitrogen are shown in parentheses for each species.
Nitrate and nitrite may be
reduced to ammonia for the purpose of nitrogen assimilation (incorporation into
organic matter, non-energy conserving) or dissimilation (using nitrate as
respiratory electron acceptor, i.e., energy-conserving, but without
incorporating the final reduced product into organic matter). Reduction of dinitrogen to ammonia is termed nitrogen fixation. All the
known enzymes catalyzing the reactions of the nitrogen cycle are
metalloenzymes. Nitrate reduction to nitrite is catalyzed by nitrate
reductases, which are molybdopterin enzymes. The
subsequent reduction of nitrite is catalyzed by two types of nitrite
reductases: those reducing nitrite to ammonia (cytochrome
c nitrite reductase, siroheme-containing
nitrite reductase) and those reducing nitrite to
nitric oxide (copper-containing nitrite reductase, cytochrome cd1 nitrite reductase)
[Einsle et al, JACS 2002, 11737; Silaghi INORG CHEM 2004, 256; Silaghi
EUR J INORG CHEM 2003, 1048]. For those nitrite reductases catalyzing nitrite
reduction to ammonia, NO and hydroxylamine are proposed to constitute reaction
intermediates, which are never released from the active site. In cytochrome c nitrite reductase,
NO is reduced to NH4+, in a mechanism proposed to involve
an {FeNO}6→{FeNO}7→{FeNO}8 sequence (cf Enemark-Feltham notation) at the lysine-ligated heme active site [Einsle et al,
JACS 2002, 11737; Silaghi EUR J INORG CHEM 2003]. On
the other hand, cytochrome cd1 nitrite reductase, which reduces nitrite to NO in vivo, can also
reduce NO to N2O, but only in vitro [Silaghi
INORG CHEM 2004]. When produced by an NO-forming nitrite reductase,
nitric oxide is further reduced to N2O by nitric oxide reductases, which, contain either cytochrome bd-or P450-type active sites [Silaghi
EUR J INORG CHEM 2003]. Nitrous oxide (a relatively inert gas) may either be
released as endproduct or further reduced to
molecular nitrogen by nitrous oxide reductases (which are multinuclear copper
proteins). Molecular nitrogen is reduced to ammonia by nitrogenases.
Thus, biological nitrate
reduction leads to one of three end-products: ammonia (which may be readily
incorporated into organic matter), nitrous oxide, or dinitrogen.
Additionally, ANAMOX, a little understood biological process is proposed to generate
molecular nitrogen from nitrate and ammonia via hydroxylamine and hydrazine.
Nitrification is an oxidative
pathway starting from ammonia and also generates partially reduced nitrogen
oxides. Thus, oxidation of ammonia to hydroxylamine is catalyzed by ammonia monooxygenase (a little understood enzyme, apparently
related to the particulate methane monooxygenase),
and is followed by oxidation of hydroxylamine to nitrite by hydroxylamine oxidase (a multiheme enzyme).
These oxidations provide electrons necessary for respiratory electron transfer
chains that facilitate lithotrophic (“rock-eating”,
i.e. extracting energy from inorganic compounds) growth. [Richardson et al, Curr. Opin.
Chem. Biol. 1999, 207]
In
addition to the above described nitrogen cycle, reduction of nitrite to
nitric oxide has also recently been proposed to be an important secondary
function of hemoglobin in humans, whereby the vasodilator molecule nitric oxide
(of Nobel Prize fame) would be generated [Gladwin, NATURE CHEM BIOL 2005, 245].
In fact, precisely due to this nitrite reductase
chemistry, nitrite is currently being introduced in clinical trials for
cardiovascular diseases in the
We
have recently proposed that linkage nitro/nitrito
isomerism is an essential part of the mechanism in the cases of copper and of heme d1-containing nitrite reductases (NIR) [Silaghi INORG CHEM 2004,
256]. Nitrite reduction by cytochrome
cd1 nitrite reductase (cd1NIR)
has long been proposed to occur (cf. Scheme 1) via N-coordination of nitrite to
the d1 heme of cd1NIR.
Protonation of a nitrite oxygen atom within the ferrous-nitrite complex would
lead to release of a water molecule, forming a weakly-bound complex, that subsequently decays via NO liberation. Nitrite
and nitric oxide adducts of the d1 heme in
cd1NIR have been characterized experimentally and computationally
[Williams et al, NATURE 1997, 406; Silaghi INORG CHEM 2004,
256; Richter et al J BIOL CHEM 2002, 3093].
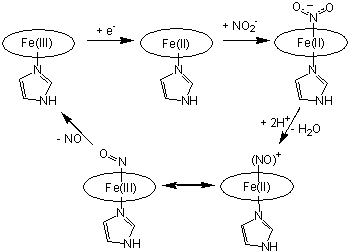
Scheme
1
Our
group has however explored an alternative possibility, involving linkage
isomerism of the nitrite at the NIR site. [Silaghi INORG CHEM 2004; Silaghi REV ROUM CHIM 2004, 496] Density functional theory results were thus reported
on the previously unexplored O-binding of nitrite to ferrous and ferric cd1NIR.
Although the N- isomer (nitro) is energetically favored over the O-nitrite (nitrito), even one single strong hydrogen bond may provide
the energy required to put the two isomers on level terms. When hydrogen
bonding existent at the cd1NIR active site was accounted for in the
computational model, the O-nitrite isomer is found to spontaneously protonate and thus yield a ferric-hydroxo
species, liberating nitric oxide. An O-nitrite ferrous cd1NIR complex appears
to be an energetically-feasible intermediate for nitrite reduction.
O-coordination would offer an advantage since the end-product of nitrite
reduction would be a ferric-hydroxo/water complex,
rather than the more kinetically inert iron-nitrosyl
complex implied by the N-nitrite mechanism. Some of this computational data is
illustrated in Figure 2.
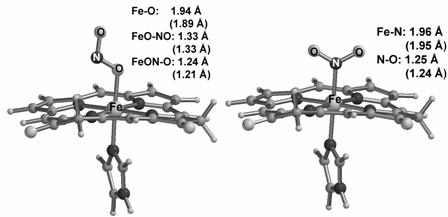
Figure 2.
Optimized geometries for O- and N- ferrous-nitrite models of
the cd1NIR active site. Distances for the corresponding
ferric-nitrite models are given in parentheses [Silaghi INORG CHEM 2004].
Our
revised catalytic cycle for cd1NIR is thus illustrated in Scheme2. This
mechanism, unlike the one in Scheme 1, reconciles for the first time the cd1NIR
chemistry with the puzzling fact that Fe(III)-NO is
kinetically inert and hence cannot possibly be a part of the cd1NIR catalytic
cycle [Silaghi INORG CHEM 2004].
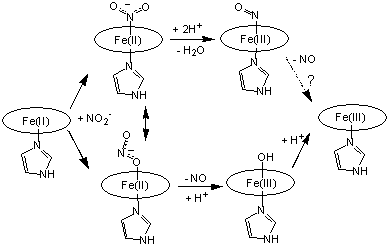
Scheme 2
The
same process as in cd1NIR, i.e. reduction of nitrite to nitric oxide, is also
catalyzed by copper-containing NIR (Cu-NIR) [Silaghi,
J INORG BIOCHEM 2006, 396; Tocheva et al, SCIENCE
2004, 867]. The proposed catalytic mechanism, illustrated in Scheme 3, has
recently been confirmed by our own computational investigations. However, we
have found that even for Cu-NIR, nitrite linkage isomerism is an intrinsic
property of the metal site, a property which the protein has to modulate in
order to achieve its goal of rapid catalytic turnover. Figure 4 illustrates
some of our computational results leading to these conclusions [Silaghi, J INORG BIOCHEM 2006, 396].
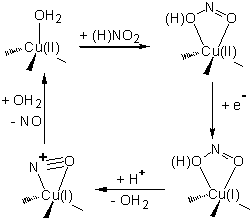
Scheme 3
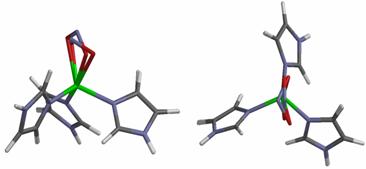
Figure 4.
Overlay (viewed along the and perpendicular to the approximate Cu-nitrite axis,
respectively) of the experimental (pdb code 1SJM) and
calculated structures of the Cu(II)-nitrite adducts. Color coding: iron, green; carbon, grey; nitrogen, blue; oxygen,
red; hydrogen, white.
Independent
experimental work has also recently supported the concept that linkage
isomerism has a profound influence on the products and mechanisms of catalytic
reduction of nitrite by free hemes and related small
complexes in solution [Kudrik et al, INORG
CHEM 2005, 6470]. In many
abiological processes, catalysis involves
redox chemistry and takes place at metal centers as
in the biological processes. The porphyrin-like
molecules and their metallocomplexes play an unique role in these processes. The huge opportunities for modification of
the porphyrin core, the strong ability of metalloporphyrinates to coordinate extra-ligands including small molecules give scientists the great
possibilities to influence the catalytic process. The additional circumstance that widen a set
of possible reaction pathways is the availability of two potential coordination
sites in some N,O-containing species. Recent studies [Kudrik et al, INORG CHEM 2003, 618] showed that reactions
of nitrate and nitrite with sodium
dithionite in the presence of CoII tetrasulfophthalocyaninate in aqueous alkaline solution
lead to different products (nitrous oxide and ammonia, respectively). These striking differences were explained in
terms of different structures of the intermediate complex between CoI phthalocyaninate
and substrate, in which nitrite and nitrate were suggested to coordinate via
nitrogen and oxygen, respectively. O-coordination of nitrite had also been
proved for ruthenium and manganese porphyrinates [Kudrik et al, INORG CHEM 2005, 6470; Silaghi INORG CHEM 2004].